 中文网站
中文网站
Nas primeiras horas do dia 29 de dezembro, o NEJM publicou online um novo estudo clínico de fase III sobre o novo coronavírus chinês VV116. Os resultados mostraram que o VV116 não foi pior que o Paxlovid (nematovir/ritonavir) em termos de duração da recuperação clínica e apresentou menos eventos adversos.

Fonte da imagem: NEJM
Tempo médio de recuperação de 4 dias, taxa de eventos adversos de 67,4%.
O VV116 é um medicamento oral nucleosídeo contra o novo coronavírus (SARS-CoV-2) desenvolvido em colaboração com a Junsit e a Wang Shan Wang Shui, e é um inibidor da RdRp juntamente com o remdesivir da Gilead, o molnupiravir da Merck Sharp & Dohme e a azelvudina da Real Biologics.
Em 2021, um ensaio clínico de fase II do VV116 foi concluído no Uzbequistão. Os resultados do estudo mostraram que o grupo tratado com VV116 apresentou melhora nos sintomas clínicos e redução significativa do risco de progressão para a forma crítica e óbito, em comparação ao grupo controle. Com base nos resultados positivos deste ensaio, o VV116 foi aprovado no Uzbequistão para o tratamento de pacientes com COVID-19 moderada a grave, tornando-se o primeiro novo medicamento coronário oral aprovado para comercialização no exterior na China [1].
Este ensaio clínico de fase III[2] (NCT05341609), liderado pelo Prof. Zhao Ren do Hospital Ruijin de Xangai, pelo Prof. Gaoyuan do Hospital Renji de Xangai e pelo Acadêmico Ning Guang do Hospital Ruijin de Xangai, foi concluído durante o surto causado pela variante Ômicron (B.1.1.529) de março a maio em Xangai, com o objetivo de avaliar a eficácia e a segurança do VV116 versus Paxlovid para o tratamento precoce de pacientes com COVID-19 leve a moderada.

Fonte da imagem: Referência 2
Um ensaio clínico multicêntrico, randomizado, controlado e com avaliação cega, envolvendo 822 pacientes adultos com Covid-19, com alto risco de progressão e sintomas leves a moderados, foi conduzido entre 4 de abril e 2 de maio de 2022 para avaliar a elegibilidade dos participantes em sete hospitais de Xangai, China. Ao final, 771 participantes receberam VV116 (384, 600 mg a cada 12 horas no dia 1 e 300 mg a cada 12 horas nos dias 2 a 5) ou Paxovid (387, 300 mg de nimatuvir + 100 mg de ritonavir a cada 12 horas por 5 dias) por via oral.
Os resultados deste estudo clínico demonstraram que o tratamento precoce com VV116 para COVID-19 leve a moderada atingiu o objetivo primário (tempo para recuperação clínica sustentada) previsto pelo protocolo clínico: o tempo mediano para recuperação clínica foi de 4 dias no grupo VV116 e de 5 dias no grupo Paxlovid (razão de risco, 1,17; IC 95%, 1,02 a 1,36; limite inferior >0,8).
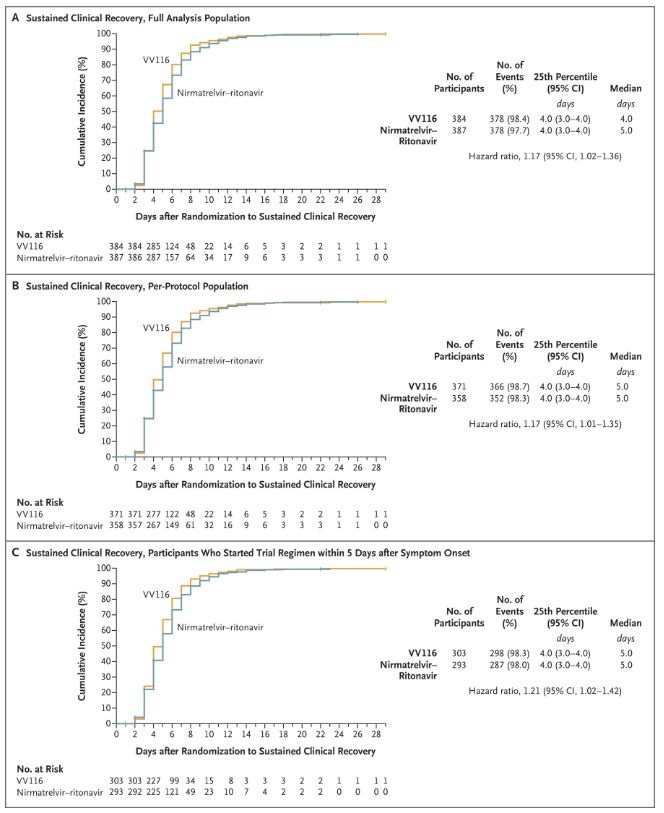
Manter o tempo de recuperação clínica
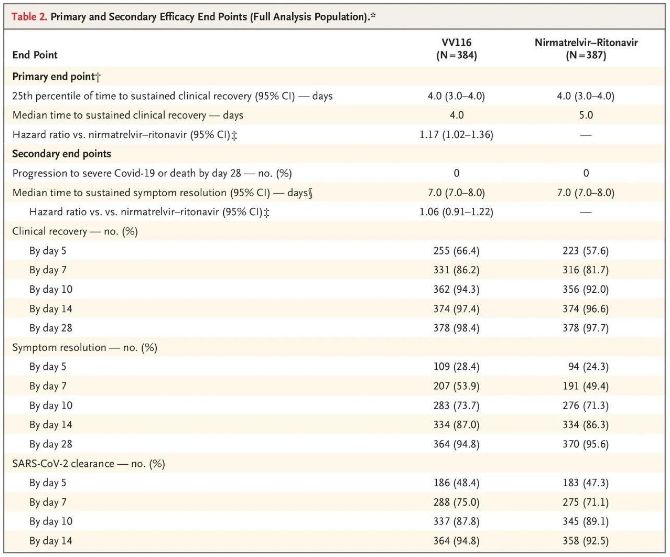
Desfechos primários e secundários de eficácia (análise abrangente da população)
Fonte da imagem: Referência 2
Em termos de segurança, os participantes que receberam VV116 relataram menos eventos adversos (67,4%) do que aqueles que receberam Paxlovid (77,3%) no acompanhamento de 28 dias, e a incidência de eventos adversos de grau 3/4 foi menor para VV116 (2,6%) do que para Paxlovid (5,7%).
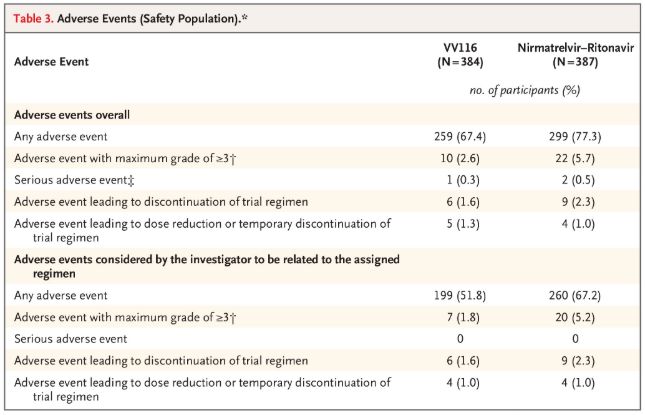
Eventos adversos (pessoas seguras)
Fonte da imagem: Referência 2
Controvérsias e perguntas
Em 23 de maio de 2022, a Juniper divulgou que o estudo clínico de Fase III para registro do VV116 versus PAXLOVID para o tratamento precoce de COVID-19 leve a moderada (NCT05341609) atingiu seu objetivo primário.

Fonte da imagem: Referência 1
Numa altura em que faltavam detalhes sobre o ensaio clínico, a controvérsia em torno do estudo de Fase III era dupla: em primeiro lugar, tratava-se de um estudo simples-cego e, na ausência de um grupo de controlo com placebo, receiava-se que seria difícil avaliar o medicamento de forma completamente objetiva; em segundo lugar, existiam dúvidas relativamente aos objetivos clínicos.
Os critérios de inclusão clínica para o estudo Juniper são: (i) resultado positivo para o teste de COVID-19, (ii) um ou mais sintomas leves ou moderados de COVID-19 e (iii) pacientes com alto risco de desenvolver COVID-19 grave, incluindo óbito. No entanto, o único desfecho clínico primário é o "tempo até a recuperação clínica sustentada".
Pouco antes do anúncio, em 14 de maio, a Juniper revisou os desfechos clínicos, removendo um dos desfechos clínicos primários, “proporção de conversões para doença grave ou morte” [3].
![]()
Fonte da imagem: Referência 1
Esses dois principais pontos de divergência também foram abordados especificamente no estudo publicado.
Devido ao surto repentino de Ômicron, a produção de comprimidos de placebo para Paxlovid não havia sido concluída antes do início do estudo e, portanto, os investigadores não puderam conduzir este estudo utilizando um desenho duplo-cego e duplo-simulado. Quanto ao aspecto simples-cego do estudo clínico, a Juniper afirmou que o protocolo foi elaborado após comunicação com as autoridades regulatórias e que o desenho simples-cego significa que nem o investigador (incluindo o avaliador do desfecho do estudo) nem o patrocinador saberão a alocação terapêutica específica do medicamento até que o banco de dados final seja bloqueado ao término do estudo.
Até o momento da análise final, nenhum dos participantes do ensaio clínico apresentou óbito ou progressão para um quadro grave de Covid-19, portanto, não é possível tirar conclusões sobre a eficácia do VV116 na prevenção da progressão para Covid-19 grave ou crítica, ou óbito. Os dados indicaram que o tempo mediano estimado desde a randomização até a regressão sustentada dos sintomas-alvo relacionados à Covid-19 foi de 7 dias (IC 95%, 7 a 8) em ambos os grupos (razão de risco, 1,06; IC 95%, 0,91 a 1,22) [2]. Não é difícil explicar por que o desfecho primário de "taxa de conversão para doença grave ou óbito", originalmente definido antes do término do ensaio clínico, foi removido.
Em 18 de maio de 2022, a revista Emerging Microbes & Infections publicou os resultados do primeiro ensaio clínico do VV116 em pacientes infectados com a variante Omicron [4], um estudo de coorte aberto e prospectivo com 136 pacientes internados confirmados.
Os dados do estudo mostraram que pacientes com infecção por Ômicron que utilizaram VV116 dentro de 5 dias após o primeiro teste de ácido nucleico positivo apresentaram um tempo de regressão do ácido nucleico de 8,56 dias, inferior aos 11,13 dias observados no grupo controle. A administração de VV116 a pacientes sintomáticos dentro do período de tempo deste estudo (2 a 10 dias após o primeiro teste de ácido nucleico positivo) reduziu o tempo de regressão do ácido nucleico em todos os pacientes. Em termos de segurança do medicamento, não foram observados efeitos adversos graves no grupo tratado com VV116.
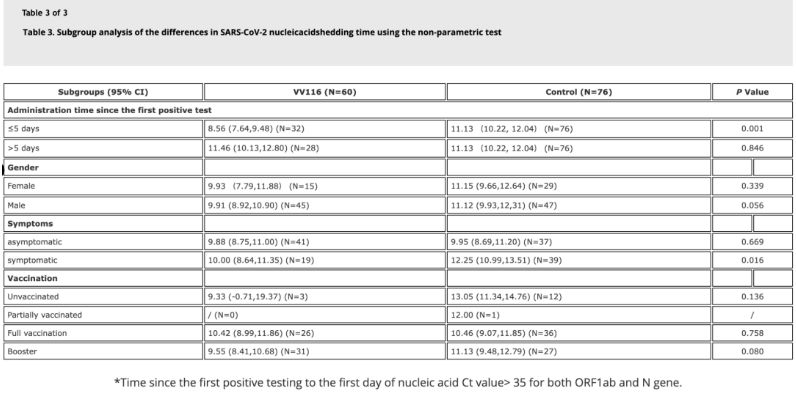
Fonte da imagem: Referência 4
Existem três ensaios clínicos em andamento com a vacina VV116, dois dos quais são estudos de fase III para COVID-19 leve a moderada (NCT05242042, NCT05582629). O outro ensaio, para COVID-19 moderada a grave, é um estudo clínico internacional multicêntrico, randomizado, duplo-cego, de fase III (NCT05279235), que visa avaliar a eficácia e a segurança da VV116 em comparação ao tratamento padrão. De acordo com o anúncio da Juniper, o primeiro paciente foi recrutado e recebeu a dose em março de 2022.

Fonte da imagem: clinicaltrials.gov
Referências:
[1]Junshi Biotech: Anúncio sobre o objetivo principal do estudo clínico de Fase III registrado de VV116 versus PAXLOVID para o tratamento precoce de COVID-19 leve a moderada
[2]https://www.nejm.org/doi/full/10.1056/NEJMoa2208822?query=featured_home[3]https://clinicaltrials.gov/ct2/show/record/NCT05341609[4] Ensi Ma, Jingwen Ai, Yi Zhang, Jianming Zheng, Xiaogang Gao, Junming Xu, Hao Yin, Zhiren Fu, Hao Xing, Li Li, Liying Sun, Heyu Huang, Quanbao Zhang, Linlin Xu, Yanting Jin, Rui Chen, Guoyue Lv, Zhijun Zhu, Wenhong Zhang, Zhengxin Wang. (2022) Perfil de infecções por Omicron e estado de vacinação entre 1.881 receptores de transplante de fígado: uma coorte retrospectiva multicêntrica. Micróbios e infecções emergentes 11:1, páginas 2636-2644.
Data da publicação: 06/01/2023
 Configurações de privacidade
Configurações de privacidade